


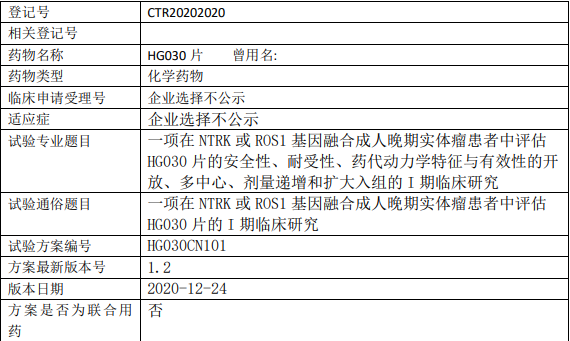
HG030临床试验,一项在NTRK或ROS1基因融合的双靶点抑制剂成人晚期实体瘤患者中评估HG030片的I期临床研究
1、试验目的
Ia 期(剂量递增阶段)
主要研究目的:评价 HG030 片在成人晚期实体瘤患者中的安全性和耐受性,确定可能出现的最大耐受剂量(MTD)。
次要研究目的:评价单次和多次口服 HG030 片在体内的药代动力学特征;确定用于 Ib 期的推荐剂量;初步评价 HG030 片在成人晚期实体瘤患者的疗效。
Ib 期(剂量扩展阶段)
主要研究目的:评价 HG030 片在 NTRK 或 ROS1 基因融合阳性成人晚期实体瘤患者中的疗效。
次要研究目的:评价 HG030 片在 ROS1 或 NTRK 基因融合阳性或其他NTRK 基因改变阳性成人晚期实体瘤患者中的安全性; 确定 II 期推荐剂量(RP2D);进一步评价多次口服 HG030 片在体内的药代动力学特征;初步评价HG030 片在其他 NTRK 基因改变(基因融合除外)阳性成人晚期实体瘤患者中的疗效;评价 ctDNA 与疗效的相关性。
2、试验设计
试验分类:安全性和有效性
试验分期:I 期
设计类型:单臂试验
随机化:非随机化
盲法:开放
试验范围:国内试验
3、受试者信息
年龄:18 岁(最小年龄)至 75 岁(最大年龄)
性别:男+女
健康受试者:无
入选标准
1.所有受试者或法定代理人必须在开始任何筛选程序之前,亲自书面签署伦理委员会批准的知情同意书。
2.年龄在 18~75 岁之间(包括界值),男女不限。
3.标准治疗后发生疾病进展或不适合/不耐受标准治疗或拒绝接受标准治疗或无标准治疗的局部晚期或转移性实体瘤受试者。
4.受试者可提供存档肿瘤组织样本或可进行新鲜肿瘤样本活检,
Ia 期:NTRK 基因或 ROS1 基因改变不作为受试者入组的必要条件;
Ib 期:包含 3 个扩展队列,需满足以下条件之一:
①NTRK 基因融合队列,经中心实验室组织学检测 NTRK 基因融合阳性者;
②ROS1 基因融合队列,克唑替尼标准治疗后疾病进展或不适合/不可耐受或拒绝克唑替尼治疗,经中心实验室组织学检测 ROS1 基因融合阳性者;
③ 其他 NTRK 基因改变队列,经中心实验室组织学检测其他 NTRK 基因改变【如,基因拷贝数变异(CNV)、点突变等】阳性者。
5.ECOG 评分 0~1 分(包括 1 分)。
6.预计生存期不少于 12 周。
7.实体瘤根据 RECIST 1.1 标准,筛选期至少存在一个可测量病灶。颅内原发肿瘤根据 RANO 标准,筛选期至少存在一个双维度可测量的病灶(经磁共振成像[MRI]证实,并可通过 RANO 标准评估),且在多成像切片上显示每个维度至少有 1 个可测量的病灶直径为1cm。
8.受试者应该有足够的器官和骨髓功能,且筛选血液学指标前7天内未接受过重组粒细胞刺激因子(长效粒细胞集落刺需要间隔2 周)、促红细胞生成素、血小板生成素、血小板生成素受体激动剂、重组白介素-11 等促进造血的药物及血小板、红细胞输注:中性粒细胞绝对计数(ANC)≥1.5×10^9/L; 血小板计数(PLT)≥100×10^9/L; 血红蛋白(Hb)≥90 g/L; 丙氨酸氨基转移酶(ALT)或天门冬氨酸氨基转移酶(AST)≤2.5倍正常值上限(ULN),肝癌或肝转移癌时 ALT 或 AST≤5 倍 ULN; 总胆红素≤1.5 倍 ULN(Gilbert 综合症受试者的总胆红素应<3 倍 ULN); 肌酐清除率≥30 mL/min; 国际标准化比值(INR)≤1.5×ULN,活化部分凝血酶时间(APTT)≤1.5×ULN。
9.男性或女性受试者,需满足以下任一条件:
无生育能力女性,特指曾做过子宫切除术、或双侧卵巢切除术、或有双侧输卵管结扎或是绝经后(停经时间≥1 年);
育龄期女性,入组前 7 天血清妊娠试验为阴性,在整个研究期间和用药结束 3 个月内都采用有效的避孕措施,例如双重屏障式避孕方法,避孕套,口服或注射避孕药物,宫内节育器等;
男性受试者,已切除输精管或同意
在研究治疗阶段以及研究药物末次给药至少 3 个月内使用避孕措施。
10.能够理解并按计划进行访视、治疗、实验室检查以及其他的研究程序。
排除标准
1.接受过除克唑替尼以外的其他ROS1-TKI或TRK-TKI的治疗或临床试验。
2. 首次研究药物治疗前 2 周或 5 个半衰期内(以时间长者为准)接受过化疗(对于亚硝基脲、丝裂霉素 C 和脂质体阿霉素,为前6周内接受过治疗)、放疗、小分子靶向治疗、免疫疗法以及首次接受研究药物治疗前 4 周内接受过抗体导向治疗者。
3. 首次研究药物治疗前,任何抗肿瘤治疗产生的不良事件未消退至或未恢复到 0 或者 1 级水平(脱发除外)(CTCAE 5.0)。
4. 研究药物治疗前 4 周内接受过外科手术,目前还未恢复者,但研究者判断不影响参加试验的小手术(例如:拔牙等)以及诊断性手术除外。
5. 目前活跃的第二恶性肿瘤,除充分治疗的包括但不限于基底或鳞状细胞皮肤癌和/或子宫颈原位癌和/或局限期前列腺癌和/或浅表膀胱癌。患有其他癌症的患者必须已无疾病 5 年或以上。
6. 存在原发性、转移性中枢神经系统肿瘤,以下情况除外:使用稳定剂量类固醇[≤4 mg/天地塞米松或同等剂量其他激素)维持或减量 2 周以上无症状者,或不需要类固醇治疗者。
7. 有临床意义的心、脑及血管异常:
例如美国纽约心脏病协会(NYHA)分级为 2 级及以上的心力衰竭,心脏左室射血分数(LVEF)<50%;
既往 6 个月内的心肌梗死、不稳定的(≥2 级,CTCAE 5.0)心律失常或不稳定的心绞痛、脑血管意外或短暂性缺血发作、肺栓塞、深静脉血栓形成;
低血压或高血压(≥3 级,CTCAE 5.0),各种有临床意义的心律、传导、静息 ECG 形态异常,包括但不限于完全性左束支传导阻滞、心房颤动/扑动、III 度传导阻滞、II度传导阻滞、PR 间期>220 msec。
8. 筛选期,在静息状态下校正的 QT 间期(Fridericia 公式校正的 QT 间期,QTcF)结果异常者:首次检测异常者需进行复测,合计 3 次,3 次心电图检查的平均 QTcF>440 msec 者。
9. 存在可能增加 QTc 延长风险或心律失常事件风险的各种因素:低钾血症、长 QT 间期综合症(LQTS),家族史中一级亲属有 LQTS或不到 40 岁就不明原因猝死,以及开始研究药物前 2 周内使用过可能延长 QT 间期的药物。
10. 已知患间质性肺病、间质纤维化、酪氨酸激酶抑制剂或其他等引起的肺部疾病。
11. 入组前不能通过引流或其他方法控制稳定时间小于 4 周的浆膜腔积液(如心包积液、胸腔积液和腹水等)
12. 其他重度或未控制疾病,研究者认为不适合参加本临床试验或会影响受试者对研究方案依从性的情况:例如严重呼吸系统疾病,如间质性肺疾病、重度哮喘等;未控制的糖尿病(经积极的干预治疗后,空腹血糖>10.0 mmol/L)。
13. 活动性易出血体质;活动性感染者,如人类免疫缺陷病毒抗体(HIVAb)阳性、梅毒抗体阳性、活动性乙型肝炎病毒(HBV)或丙型肝炎病毒(HCV)感染。活动性乙型肝炎定义为:乙肝核心抗体(HBcAb)或乙肝表面抗原(HBsAg)阳性,且 HBV DNA ≥ 2000IU/mL(相当于 104 拷贝/mL);活动性丙型肝炎定义为 HCV RNA高于检测下限。
14. 任何影响受试者吞服药物或口服吸收障碍的情况,包括但不限于:严重慢性胃肠道疾病、接受过胃肠道手术、肠梗阻、活动性消化道溃疡、难治性恶心或呕吐症状等。
15. 周围神经病变 2 级以上者。
16. 有药物滥用史或酒精滥用史或患有精神疾病,或怀疑对研究药物或其任何成分过敏或不耐受者。
17. 首次研究药物治疗前 2 周内使用过中度或强效 CYP3A4 抑制剂或诱导剂。
18. 2 个月内参加过其他药物或医疗器械的临床试验或在其他试验药物代谢 5 个半衰期时间内。
19. 研究者判断不适合进入本研究的其他情况。
研究者信息
1、主要研究者信息
姓名:沈琳
学位:医学博士
职称:教授
单位名称:北京大学肿瘤医院
2、各参加机构信息
| 序号 | 机构名称 | (主要)研究这 | 国家 | 省(州)-城市 |
| 1 | 北京大学肿瘤医院 | 沈琳 | 中国 | 北京市-北京市 |
| 2 | 四川大学华西医院 | 王永生 | 中国 | 四川省-成都市 |
| 3 | 河南省肿瘤医院 | 赵艳秋 | 中国 | 河南省-郑州市 |






















 首页
首页 咨询
咨询 方舟新药
方舟新药 营养商城
营养商城