

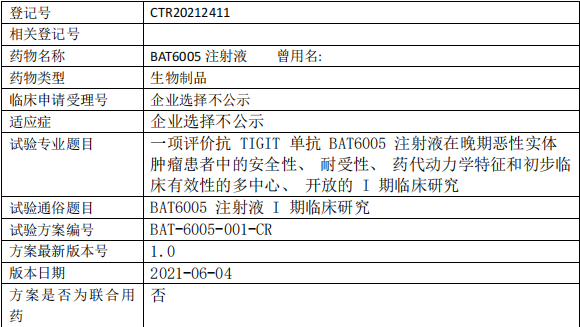
百奥泰生物BAT6005临床试验,TIGIT单抗BAT6005注射液晚期恶性实体肿瘤的I期临床试验
1、试验目的
主要目的
·评估 BAT6005 注射液治疗局部晚期或转移性实体瘤患者的安全性与耐受性;
·探索最大耐受剂量(MTD) 或最大给药剂量(MAD) 并为 II 期或后续临床 研究提供推荐剂量及合理的给药方案。
次要目的
·评价 BAT6005注射液在局部晚期或转移性实体瘤患者中单次给药和多次给 药的药代动力学(PK) 特征;
·评价 BAT6005 注射液的免疫原性;
·评价 BAT6005 注射液的药效学特性;
·初步评价 BAT6005 注射液的抗肿瘤疗效
2、试验设计
试验分类:安全性和有效性
试验分期:I 期
设计类型:单臂试验
随机化:随机化
盲法:开放
试验范围:国内试验
3、受试者信息
年龄:18 岁(最小年龄)至无上限(最大年龄)
性别:男+女
健康受试者:无
入选标准
1.年龄: ≥18 周岁, 性别: 男女不限;
2.研究者评估预期生存期至少为 3 个月;
3.美国东部肿瘤协作组(ECOG)体能状态评分要求为 0 或 1 分;
4.尚无标准治疗、 或标准治疗失败、 或标准治疗不适用的, 经组织学或细 胞学确诊的局部晚期或转移性恶性实体肿瘤患者;
5.根据肿瘤疗效评估标准 RECIST 1.1, 剂量递增阶段须有可评价的肿瘤病 灶, 剂量扩展阶段须至少存在一个可测量的肿瘤病灶;
6.具备足够的器官和骨髓储备功能, 定义如下: 系统 实验室参考值 血常规(首次给药前 14 天内未输血、 未使用造血刺激因子以及未使用药物纠正血细 胞数) 中性粒细胞绝对计数(ANC)≥1.5× 109 /L 血小板计数 ≥75× 109/L 血红蛋白 ≥85g/L凝血功能 凝血酶原时间(PT) 或国际标准 化比值(INR)和活化部分凝血 活酶时间(APTT) ≤ 1.5× ULN(未接受抗凝治疗者) 接受口服抗凝药物治疗且 INR 在 2~3 者可纳入肝功能总胆红素(TBIL) — ≤1.5× ULN 肝细胞癌、 Gilbert’ s 综合征、 肝 转移 ≤2× ULN 丙氨酸氨基转移酶(ALT) 、 天—≤2.5× ULN 方案编号: BAT-6005-001-CR 版本号 1.0 版本日期 2021-06-04 第 10 页 共 82 页 冬氨酸氨基转移酶(AST)肝细胞癌、 肝转移 ≤5× ULN 肾功能 血清肌酐清除率 >50ml/min(采 用 Cockcroft-Gault 公 式 , 见 附 录)
7.具备生育能力的女性患者, 必须在首次给药前 7 天内血清妊娠试验阴性 并且愿意在研究期间直至研究最后一次给药后 6 个月内采取有效的节育/ 避孕方法防止妊娠。 男性患者必须同意在研究期间直至研究最后一次给药后 6 个月内, 采取有效的避孕方法; 绝经后的妇女必须闭经至少 12 个月才被认 为不具备生育能力;
排除标准
1.既往曾接受过抗TIGIT单克隆抗体或具有抗TIGIT活性的双抗等药物治疗;
2. 在首次使用研究药物前 4 周内接受过化疗、放疗、生物治疗、内分泌治 疗、 免疫治疗等抗肿瘤治疗, 除外以下几项:
· 亚硝基脲或丝裂霉素 C 为首次使用研究药物前 6 周内;
· 口服氟尿嘧啶类和小分子靶向药物为首次使用研究药物前 2 周或药物的 5 个半衰期内(以时间长的为准) ;
· 有抗肿瘤适应症的中药为首次使用研究药物前 2 周内
3. 在首次使用研究药物前 4 周内接受过其它未上市的临床研究药物或治疗;
4. 筛选前4周内接种过或计划在研究期间内接种活/减毒疫苗以及mRNA 疫苗;
5. 孕妇或哺乳期妇女;6. 既往抗肿瘤治疗所致 AE 未恢复至 CTCAE 5.0≤ 1 级的患者;(研究者判 断无安全风险的毒性除外,如脱发、 2级外周神经毒性、经激素替代治疗稳 定的甲状腺功能减退等) ;
7. 具有临床症状的脑实质转移或脑膜转移,经研究者判断不适合入组者;
8. 在首次使用研究药物前 4 周内接受过主要脏器外科手术(不包括穿刺活检)或出现过显著外伤,或需要在试验期间接受择期手术的患者;
9. 具有组织或器官移植手术史者;
10. 首次给药前伴有活动性感染,且目前需要静脉抗感染治疗者;
11. 已知有人类免疫缺陷病毒(HIV) 感染史
12. 活动性乙型肝炎;
注: 符合下列标准的乙肝受试者也可不排除:
a.首次给药前 HBV 病毒载量必须<500 IU/ml 或低于所在研究中心的检 测下限(仅当所在研究中心的检测下限高于500IU/ml时),研究者需 考虑在研究期间给予抗 HBV 治疗;
b.对于抗 HBc(+) 、 HBsAg(-) 、 抗 HBs(-) 和 HBV 病毒载量(-) 的受试者,不需要接受预防性抗 HBV 治疗, 但是需要密切监测病毒 再激活;
13. 活动性的 HCV 感染受试者(HCV 抗体阳性且 HCV-RNA 水平高于检测下限) ;
14. 未经治疗或正在治疗的结核病受试者, 包括但不仅限于肺结核; 经规范 抗结核治疗并经研究者确认已治愈者可纳入;
15. 已知有严重过敏史, 或已知受试者既往对大分子蛋白制剂/单克隆抗体曾 发生过≥3 级过敏反应者;
16. 患有活动性、 或曾患过且有复发可能的自身免疫性疾病的患者(如系统 性红斑狼疮、 类风湿性关节炎、 血管炎等) , 除外临床稳定的自身免疫甲状 腺疾病、 I 型糖尿病患者;
17. 在首次使用研究药物前 14 天内接受过全身使用的糖皮质激素(强的松 > 10mg/天或等效剂量的同类药物) 或其他免疫抑制剂治疗; 除外以下情 况: 使用局部、 眼部、 关节腔内、 鼻内和吸入型糖皮质激素治疗, 短期使用 糖皮质激素进行预防治疗(例如预防造影剂过敏) 。
18. 既往出现过≥3 级 irAE;
19. 当前存在间质性肺疾病者;
20. 有严重的心脑血管疾病史, 包括但不限于:
· 有严重的心脏节律或传导异常, 如需要临床干预的室性心律失常、 Ⅱ-Ⅲ度房室传导阻滞等;
· 首次给药前 6 个月内发生急性冠脉综合征、 充血性心力衰竭、 主动脉夹层、 脑卒中或其他 3 级及以上心脑血管事件;
· 存在美国纽约心脏病协会(NYHA)心功能分级≥II 级的心力衰竭或左室射血分数(LVEF)<50%·临床无法控制的高血压(本方案定义为虽然采用抗高血压治疗,但治疗后仍收缩压>150mmHg和/或舒张压>100mmHg) ;
21. 临床无法控制的第三间隙积液, 经研究者判断不适合入组;22. 已知有精神类药物滥用或吸毒史, 并被认为会影响本研究依从性的患者;
23. 研究者认为不适合参与本研究的患者。
4、研究者信息
1.主要研究者信息
姓名:李进
学位:博士
职称:主任医师
单位名称:上海市东方医院
2.各参加机构信息
| 序号 | 机构名称 | (主要)研究者 | 国家 | 省(州)-城市 |
| 1 | 上海市东方医院 | 李进 | 中国 | 上海市-上海市 |
| 2 | 山东省肿瘤医院 | 李因涛 | 中国 | 山东省-济南市 |






















 首页
首页 咨询
咨询 方舟新药
方舟新药 营养商城
营养商城