

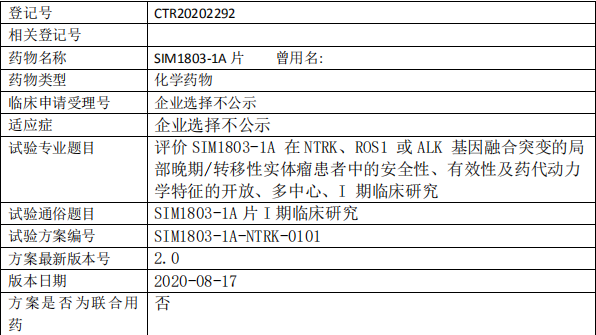
SIM1803-1A临床试验,ROS1、ALK、NTRK临床试验,SIM1803-1A片I期临床研究
1、试验目的
Ia 期-主要目的:评价 SIM1803-1A 的安全性和耐受性;评价 SIM1803-1A 的药代动力学特征;确定 SIM1803-1A 的最大耐受剂量(MTD)和后续研究的合适推荐剂量(RD)。
次要目的:初步评价 SIM1803-1A 治疗 NTRK、ROS1 或 ALK 基因融合突变的局部晚期/转移性实体瘤患者的有效性。
Ib 期-主要目的:评价SIM1803-1A 治疗 NTRK、ROS1 或 ALK 基因融合突变的局部晚期/转移性实体瘤患者的有效性。
次要目的:评价 SIM1803-1A 治疗 NTRK、ROS1 或 ALK 基因融合突变的局部晚期/转移性实体瘤患者的安全性;评价Ⅰa 期推荐剂量下 SIM1803-1A的药代动力学特征。
2、试验设计
试验分类:安全性和有效性
试验分期:I 期
设计类型:单臂试验
随机化:非随机化
盲法:开放
试验范围:国内试验
3、受试者信息
年龄:18 岁(最小年龄)至无上限(最大年龄)
性别:男+女
健康受试者:无
入选标准
1.自愿参加临床试验,并签署知情同意书;
2.年龄 18 周岁以上(含),性别不限;
3.经组织学和/或细胞学确诊的局部晚期/转移性实体瘤患者;
4.晚期肿瘤疾病患者接受系统性标准治疗(根据各个瘤种的指南)后,标准治疗失败或无法耐受;
5.剂量扩展阶段需既往检测存在 NTRK、ROS1 或 ALK 基因融合突变;注:可以接受经 FDA CAP 或 CLIA 认证的实验室或经中国临检中心认可的当地实验室的基因检测结果。
6.同意提供肿瘤组织用于 NTRK、ROS1 或 ALK 等基因分析(无法提供足够肿瘤组织进行基因分析的患者仅在研究者与申办方医学监察员商量,且得到书面同意的情况下方可入组);
7.ECOG 评分 0~1;
8.预期生存时间≥3 个月;
9.主要器官功能基本正常,筛选期实验室检查值符合下列标准:
中性粒细胞绝对计数>1.5 ×109/L,血小板>100×109/L,血红蛋白>100g/L 肌酐清除率(CrCl)或肾小球滤过率(GFR)(roft-Gault 公式)>60 mL/min 总胆红素(血清)<1.5 × ULN,血胆红素≥1.5 × ULN 的 Gilbert 病患者可在申办方同意后入组,AST 及 ALT<3 × ULN 国际标准化比值(INR)或部分凝血酶原时间(aPTT)<1.5 × ULN,除非受试者接受抗凝治疗
10.筛选期育龄期女性受试者的血清妊娠试验检查结果呈阴性;
11.受试者同意自签署知情同意书至最后末次给药后90 天内使用可 靠的避孕方法。包括但不限于:禁欲、男性接受输精管切除术、女性绝育手术、有效的宫内节育器、有效的避孕药物。注:非育龄期女性包括永久绝育(子宫切除术、双侧卵巢切除术或双侧输卵管切除术)和已绝经。对于≥50 岁女性,如果在计划入组日期前 12 个月已停经且无其他医学病因,则认为该名女性已绝经。对于<50 岁的女性,如果停止外源性激素治疗后已停经 12 个月或以上、且促卵泡生成激素(FSH)水平在绝经后范围内,则认为该名女性已绝经。
排除标准
1.首次给药前 6 个月内患有具有临床意义的心脑血管疾病,经研究者判断可能会妨碍受试者全程参与研究的患者;
2. 首次给药前 6 个月内心电图示 QTcF 间期>480ms;
3. 有症状未控制的原发性中枢神经系统肿瘤或脑转移,或任意症状的脑膜转移
4. 已知的肺间质疾病、肺间质纤维化或既往有酪氨酸激酶抑制剂导致的肺炎病史,放疗所引起的肺炎除外;
5. 周围神经病变≥2 级
6. 患有吞咽困难或者影响药物吸收的胃肠道疾病(比如克罗恩病,溃疡性结肠炎,或短肠综合征)或其他吸收不良情况;
7. 首次给药前,尚未从任何既往治疗引起的不良事件和/或并发症中恢复至正常或≤1 级;脱发(任何级别)的患者可以入组;
8. 首次给药前 3 周内接受过已上市的系统性抗肿瘤治疗,包括化疗、大分子单抗、免疫治疗等;首次给药前 2 周内或药物的 5 个半衰期内(以时间长的为准)接受过小分子靶向药或小分子内分泌治疗治疗;首次给药前 2 周内接受过有抗肿瘤适应症的中/草药;
9. 首次给药前 4 周内接受过除诊断或活检外的其他重大手术;
10. 首次给药前 2 周内接受过针对胸部以外部位放疗或是首次给药前 6 个月内接受过针对胸部放疗;
11. 首次给药前 4 周内或已知的研究药物的 5 个半衰期(以时间短的为准)内作为受试者参加其它临床试验(筛选失败除外);
12. 首次给药前 2 周内使用过 CYP3A4 强抑制剂或强诱导剂;
13. 严重的感染需要静脉输注抗生素或住院治疗
14. 未控制的乙型肝炎病毒感染(HBsAg 阳性),或丙型肝炎病毒感染(抗 HCV 阳性),或人类免疫缺陷病毒 HIV 感染或活动性梅毒感染; 乙型肝炎患者,若 HBV DNA 的 PCR 检测结果为阴性则可入组,但需要在研究治疗期间接受预防性抗病毒治疗,并且需要每周期进行 HBV DNA 的 PCR 检测 丙型肝炎患者,若 HCV RNA 的PCR 检测结果为阴性则可入组;
15. 既往有严重过敏史者,或对研究药物的任何活性或非活性成分过敏者;
16. 妊娠期或哺乳期患者;
17. 研究者认为受试者存在任何临床或实验室检查异常或其他原因而不适合参加本临床研究。
研究者信息
1、主要研究者信息
姓名:陆舜
学位:医学博士
职称:正高级
单位名称:上海市胸科医院
2、各参加机构信息
机构名称:上海市胸科医院
(主要)研究者:陆舜
国家:中国
省(州)-城市:上海市 -上海市






















 首页
首页 咨询
咨询 方舟新药
方舟新药 营养商城
营养商城